
Turinys
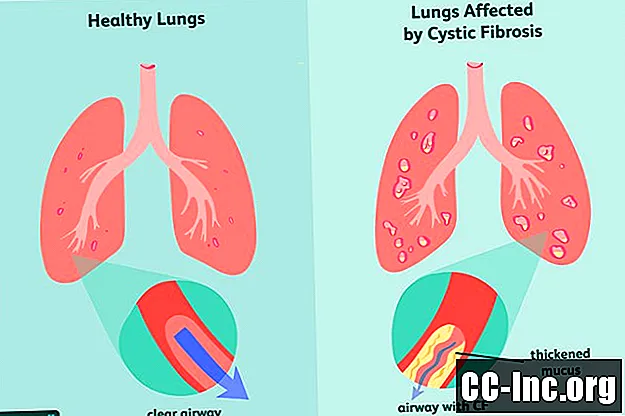
Cistinė fibrozė (CF) yra paveldimas, gyvybei pavojingas sutrikimas, pažeidžiantis plaučius ir virškinamąjį traktą. Ją sukelia defektas genas, sukeliantis sutirštėjusių gleivių gamybą, kurios užkemša kvėpavimo takus ir blokuoja virškinimo fermentų sekreciją.Simptomai yra progresuojantys ir dažnai sunkūs, jie gali apimti kvėpavimo sutrikimus, pasikartojančias plaučių infekcijas, prastą augimą, vyrų nevaisingumą ir lėtinį kasos, kepenų, inkstų ir širdies uždegimą.
CF galima diagnozuoti atliekant kraujo tyrimus, genetinę atranką ir procedūrą, vadinamą prakaito chlorido testu.
Nors nuo CF negalima išgydyti, yra gydymo būdų, kurie gali pagerinti ir gyvenimo trukmę, ir kokybę.
Tai apima kvėpavimo takų šalinimo būdus, įkvepiamus antibiotikus, gleivių skiediklius, kasos fermentus, kaloringą dietą ir naujesnės kartos vaistus, vadinamus CFTR moduliatoriais. Sunkiais atvejais gali prireikti plaučių transplantacijos.
Cistinės fibrozės simptomai
Kaip genetinis sutrikimas, cistinė fibrozė yra kažkas, su kuo gimstate. Gimimo metu jis gali ir nebūti, ir gali prireikti mėnesių ar net metų, kol atsiras jokių ligos požymių. Iki to laiko plaučiai ir virškinamasis traktas jau gali patirti žalą, kurios negalima panaikinti.
Dažniausiai pasitaikantys ankstyvieji CF požymiai ir simptomai yra šie:
- Pirmojo kūdikio išmatų (meconiumo) užsikimšimas
- Druskingo skonio oda
- Lėtinis kosulys, švokštimas ar spalvoti skrepliai
- Laisvos, riebios ir paprastai kvapo išmatos
- Plaučių infekcija, dažnai pasikartojanti
- Blogas augimas ir nesugebėjimas klestėti
Jei šių simptomų nepavyksta suvaldyti, stresas plaučiuose (ir nesugebėjimas priaugti svorio) gali turėti kumuliacinį poveikį, paveikti kelis organus ir padidinti ligos komplikacijų riziką.
Kai kurios būdingesnės komplikacijos yra:
- Vėlyvas brendimas
- Bronchektazė (lėtinis plaučių sienelių sustorėjimas)
- Svorio metimas
- Pankreatitas (kasos uždegimas)
- Vyrų nevaisingumas
- Plaučių hipertenzija (padidėjęs kraujospūdis plaučiuose)
- Tulžies pūslės akmenys
- Su cistine fibroze susijęs diabetas
- Cor pulmonale (dešinysis širdies nepakankamumas)
- Cirozė (funkcinis kepenų randėjimas)
Kadangi CF sukelia progresuojantį ląstelių ir audinių pažeidimą, bet kokia plaučių ir kitų organų padaryta žala bus iš esmės negrįžtama. Mirtis dažniausiai pasireiškia kvėpavimo nepakankamumu, po to - širdies ir kepenų nepakankamumu.
Cistinės fibrozės simptomai
Priežastys
Cistinę fibrozę sukelia cistinės fibrozės transmembraninio receptoriaus (CFTR) geno, kuris yra atsakingas už CFTR baltymo gamybą, mutacija. Tai yra baltymas, kurio organizmas turi reguliuoti druskos ir vandens srautą ląstelėse ir iš jų . Jei baltymas yra deformuotas ar sugedęs, jis gali sukelti dehidrataciją ląstelės paviršiuje, o tai gali sutirštinti aplinkines gleives.
CF yra autosominis recesyvinis sutrikimas, o tai reiškia, kad jums reikia paveldėti CFTR mutaciją tiek iš motinos, tiek iš tėvo, kad sirgtumėte šia liga. Jei paveldėsite tik vieną sugedusį geną, neturėsite CF, bet būsite mutavusio geno nešėjas.
Jūs galite paveldėti ligą, jei kiekvienas iš jūsų tėvų turi CFTR mutaciją arba pačią CF. Jei abu tėvai yra vežėjai, turėtumėte:
- 25 procentų tikimybė turėti CF
- 50 procentų tikimybė būti vežėju
- 25 proc. Tikimybė, kad tai nebus paveikta
Kita vertus, jei vienas iš jūsų tėvų yra vežėjas, o kitas - CF, turite 50/50 tikimybę, kad turėsite CF arba būsite vežėjas.
Cistinė fibrozė yra viena iš labiausiai paplitusių genetinių ligų, kuria serga maždaug vienas iš 2500 JAV gimusių kūdikių.
Dažniausiai jis paplitęs tarp kaukaziečių ir ispanų, rečiau pasitaiko afrikiečių ar azijiečių kilmės žmonėms.
Cistinės fibrozės rizikos veiksniaiDiagnozė
Cistinei fibrozei diagnozuoti naudojami keli tyrimai. Jie veikia tiesiogiai nustatydami CFTR mutaciją arba netiesiogiai matuodami biologinius pokyčius, atitinkančius ligą. Diagnozės metodas gali skirtis nėštumo metu, gimus kūdikiui ar bet kada vėliau.
Cistinės fibrozės gydytojo diskusijų vadovas
Gaukite mūsų atspausdintą vadovą kitam gydytojo paskyrimui, kuris padės jums užduoti teisingus klausimus.

Iš dviejų standartinių testų, dažniausiai naudojamų CF diagnozuoti:
- Prakaito chlorido tyrimas, taip pat žinomas tiesiog kaip prakaito testas, matuoja chloro kiekį ant odos. Kadangi CF trukdo perkelti druską į ląsteles ir iš jų, prakaite bus kaupiama druska.
- Genetinis CFTR testavimas yra naudojamas nustatant dažniausiai pasitaikančias CFTR mutacijos mutacijas. Nors žinoma, kad yra daugiau kaip 2000 CFTR mutacijų, sukeliančių cistinę fibrozę, 23, įtrauktos į standartinę grupę, yra labiausiai tikėtini įtariamieji.
Nėštumo metu CFTR genetinis testas gali būti naudojamas skysčiams, gautiems atliekant amniocentezę, arba ląstelėms, ištrauktoms naudojant choriono gaurelių mėginius (CVS), tirti.
Naujagimio patikra taip pat paprastai naudojamas diagnozuoti CF ir šiandien yra įgaliotas visose 50 valstijų ir Kolumbijos apygardoje. Tai, ką tai reiškia, skirsis priklausomai nuo to, kur jūs gyvenate JAV. Jei naujagimio patikros rezultatai yra teigiami, diagnozei patvirtinti būtų naudojamas prakaito testas.
Kaip diagnozuojama cistinė fibrozėGydymas
Nors cistinės fibrozės išgydyti negalima, pažanga gydant pailgino šia liga gyvenančių žmonių gyvenimo trukmę.
CF gydymas yra keturis kartus: užkirsti kelią infekcijoms, išlaikyti plaučių funkciją, normalizuoti virškinimą ir sulėtinti ligos progresavimą.
Tarp terapinių priemonių, naudojamų CF valdyti:
- Kvėpavimo takų šalinimo būdai (ACT) atliekami norint išstumti ir išstumti iš plaučių susikaupusias gleives. Būdai apima kosulį, krūtinės mušimą ar krūtinės sienelės svyravimą.
- Riebi, kaloringa dieta yra naudojamas riebalų, baltymų ir maistinių medžiagų absorbcijos sutrikimui žarnyne kompensuoti.
- Kasos fermentų papildai yra naudojami virškinimo fermentams, kurių kasa negali gaminti dėl per didelio gleivių kaupimosi, stiprinti.
- Antibiotikai yra vartojami kasdien, kad būtų išvengta bakterinių plaučių infekcijų.
- Mukolitikai- gali būti naudojami vaistai, vartojami gleivėms skystinti prieš ACT.
- CFTR moduliatoriai yra nauja vaistų klasė, galinti ištaisyti tam tikrus CFTR baltymo defektus ir atkurti jų reguliavimo funkciją.
- Deguonies terapija gali būti naudojamas ūmių epizodų metu, kai labai sutrinka kvėpavimas.
- Enterinė mityba, taip pat žinomas kaip maitinimas mėgintuvėliu, gali būti naudojamas, jei negalite išlaikyti svorio normaliai maitindamiesi.
- Plaučių transplantacija yra laikoma, kai jūsų plaučiai nebegali palaikyti išgyvenimo be mechaninės ventiliacijos.
Įveikti
1938 m., Kai cistinė fibrozė pirmą kartą buvo priskirta ligai, vaikai retai gyveno po pirmųjų gyvenimo metų. Aštuntajame dešimtmetyje galima tikėtis gyventi 20–25 metus. Šiandien vaizdas visiškai pasikeitė žmonėms, gyvenantiems 40–50 metų, jei gydymas pradedamas anksti ir jo laikomasi.
Tai nereiškia, kad CF yra mažiau rimtas nei kada nors anksčiau. Tai įvykis, keičiantis gyvenimą, reikalaujantis kruopštumo ir nuoseklumo, kad ne tik susitvarkytumėte su liga, bet ir gyventumėte kuo aukštesnį gyvenimo lygį.
Šiuo tikslu turite normalizuoti CF savo gyvenime nustatydami tvarką ir praktiką, kad išvengtumėte peripetijų, kurios gali sukelti stresą ir padidinti negalią. Tarp svarstymų turėtumėte:
- Tvarkykite savo mitybą. Žmonėms, sergantiems CF, dažnai reikia dvigubai daugiau kalorijų nei per dieną.
- Reguliariai sportuokite. Idealiu atveju fitneso treniruotės turėtų apimti mažiausiai 20–30 minučių aerobinę veiklą tris kartus per savaitę. Rask ką nors malonaus, ką galėtum padaryti visą gyvenimą.
- Laikykite gerai hidratuotą. Tokiu būdu plaučiai ir žarnynas tinkamai veikia. Atsižvelgiant į jūsų amžių, per dieną turėtumėte išgerti ne mažiau kaip šešias aštuonias aukštas stiklines vandens.
- Teisingai atlikite kvėpavimo takų tarpą. Keičiantis jūsų sveikatai, gali pasikeisti ir jums reikalingi kliringo įrankiai. Jei nepasiekiate reikiamų rezultatų, pasitarkite su savo pulmonologu ar kineziterapeutu.
- Kreipkitės pagalbos. Be draugų ir šeimos narių, galite susisiekti su artimiausiu Cistinės fibrozės fondo (CFF) skyriumi, kad prisijungtumėte prie palaikymo tinklo jūsų vietovėje.
- Kreipkitės į finansinę pagalbą. CFF siūlo paslaugas, kurios padeda šeimoms geriau susidoroti su didelėmis CF gydymo išlaidomis.
Žodis iš „Wellwell“
Nors naujagimių patikros žymiai padidino kūdikių CF diagnozę, daugiau nei 25 proc. Diagnozių nustatoma tik vaikystėje, paauglystėje ir ankstyvame suaugusiųjų amžiuje.
Tai yra problematiška, nes ankstyva diagnostika ir gydymas gali užkirsti kelią daugeliui sunkesnių CF komplikacijų, kol nebus padaryta rimta žala. Nors gydymas negali sustabdyti ar pakeisti ligos, jis gali užtikrinti daug daugiau metų be ligų.
Šiuo tikslu svarbu žinoti ankstyvuosius CF simptomus ir pasitarti su savo gydytoju, jei įtariate, kad jūsų vaikas gali sirgti šia liga. Remiantis Viskonsino universiteto medicinos ir visuomenės sveikatos mokyklos tyrimais, tai ypač pasakytina apie valstybes, kuriose atliekamas tik IRT kraujo tyrimas, dėl kurio tiek 5 procentai vaikų gali patirti uždelstą diagnozę, tiek klaidingai neigiamą rezultatą. .
Kokių simptomų galite tikėtis sirgdami cistine fibroze?